Acute Leukaemia of Ambiguous Lineage Presenting as a Focal Bone Lesion: a Case Report
CASE REPORT
Acute Leukaemia of Ambiguous Lineage Presenting as a Focal Bone Lesion: a Case Report
H Yi1, M Dhamija2, H Dholaria3, R Kotecha2, D Roebuck3
1 Division of Paediatrics, Medical School, University of Western Australia, Australia
2 Department of Haematology and Oncology, Perth Children’s Hospital, Australia
3 Department of Medical Imaging, Perth Children’s Hospital, Australia
Correspondence: Prof. D Roebuck. Department of Medical Imaging, Perth Children’s Hospital, Australia. Email: derek.roebuck@health.wa.gov.au
Submitted: 9 Oct 2020; Accepted: 19 Jan 2021.
Contributors: HY and DR designed the study. All authors acquired the data. All authors analysed the data. HY and DR drafted the manuscript.
All authors critically revised the manuscript for important intellectual content. All authors had full access to the data, contributed to the study,
approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of Interest: All authors have disclosed no conflicts of interest.
Funding/Support: This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Data Availability: All data generated or analysed during the present study are included in this published article.
Ethics Approval: The study was approved by the institutional Research Ethics Committee (Ref 1769/EP).
INTRODUCTION
Acute leukaemia is the most common childhood
malignancy. Almost all cases are classified as acute
lymphoblastic leukaemia (ALL) or acute myeloid
leukaemia (AML). Acute leukaemia of ambiguous
lineage (ALAL) is a rare form of acute leukaemia that
cannot be classified by a single lineage.[1] Like other acute
leukaemias, ALAL typically presents with nonspecific
symptoms such as fatigue, fever, or bleeding.[2] Although
presentation with musculoskeletal symptoms such as
bone pain is not uncommon in acute leukaemia, a focal
pattern of bone involvement at presentation is rarely
observed and has not been described in children with
ALAL. We describe the case of a 16-month-old girl
with ALAL who presented with a focal destructive bone
lesion.
CASE REPORT
A 16-month-old girl was referred to our oncology unit with a 3-week history of progressive pain, irritability
and inability to bear weight on her left lower limb. She was a twin born at 34 weeks’ gestation from an in vitro
fertilisation pregnancy and was well prior to presentation.
A radiograph of the left femur (not shown) revealed a permeative lytic lesion in the left distal femoral
metaphysis. Lower limb computed tomography (CT)
showed a destructive lesion, with a small soft tissue
mass, suggestive of a primary bone tumour, Langerhans
cell histiocytosis, or a metastasis (Figure 1). Magnetic
resonance imaging (MRI) of the thighs showed
numerous other focal bone lesions, all asymptomatic
(Figure 2), and CT of the chest and abdomen showed no
evidence of lymph node enlargement, organomegaly or
soft tissue mass. Initial blood investigations and urinary
catecholamines were normal. The femoral lesion was
biopsied, revealing a largely necrotic undifferentiated
neoplasm with focal areas of viable tissue and large
round blue cells. The viable cells were positive for
CD99, CD43, CD34, CD117, vimentin and CD56, and
negative for lymphoid and other solid tumour markers
on immunohistochemical staining.
Figure 1. Coronal computed tomography image of distal left
femur showing a destructive metaphyseal lesion with a permeative
growth pattern, subperiosteal new bone formation (thin arrow) and
a soft tissue mass (thick arrow).
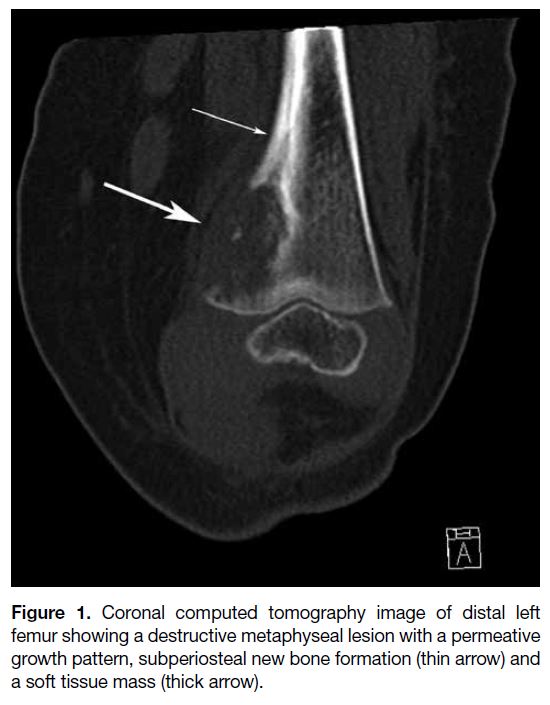
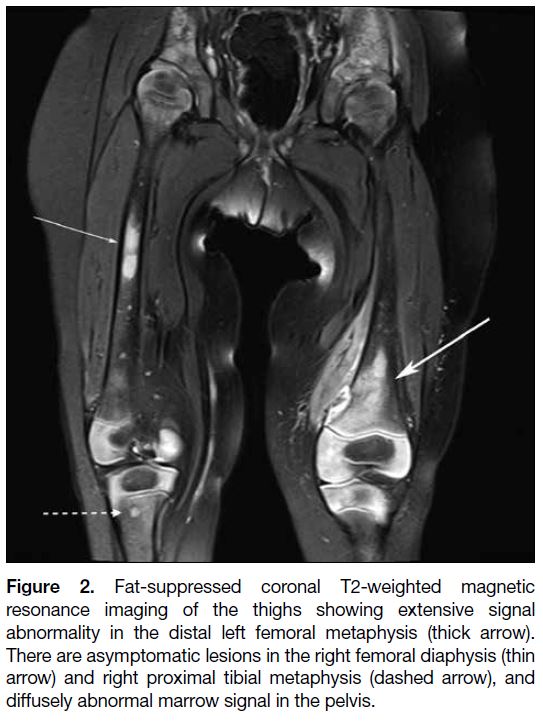
Figure 2. Fat-suppressed coronal T2-weighted magnetic
resonance imaging of the thighs showing extensive signal
abnormality in the distal left femoral metaphysis (thick arrow).
There are asymptomatic lesions in the right femoral diaphysis (thin
arrow) and right proximal tibial metaphysis (dashed arrow), and
diffusely abnormal marrow signal in the pelvis.
During her diagnostic workup, she developed
left-sided facial weakness with drooling. MRI
demonstrated extensive calvarial marrow infiltration,
with compression of the facial nerve by a left mastoid
lesion, and extensive vertebral infiltration with
pathological fractures. Cerebrospinal fluid examination
was negative for malignant cells. Positron emission
tomography–computed tomography (PET-CT) with
18F-fluorodeoxyglucose revealed diffuse skeletal
and splenic uptake. She was started on emergency
chemotherapy with carboplatin and etoposide while
awaiting confirmation of the final diagnosis.
The bone marrow aspirates and trephines revealed up to 20% blasts, with immunohistochemical staining
showing the same characteristics as the bone biopsy.
Cytogenetic assessment of the bone marrow aspirates
revealed a 46,XX,der(7)t(7;16)(q36;p11.2),der(8)t(1;8)
(q25;p23),der(16)inv(16)(p11.2q24)t(7;16)(q36;p11.2)
[9]/46,XX[11] karyotype consistent with a neoplastic
clone. Bone biopsies from her left iliac wing and tibia
showed significant blast infiltration, strongly positive for
CD33, CD34, CD117, CD16/56 and cytoplasmic CD3,
but negative for cytoplasmic myeloperoxidase, HLA-DR,
nuclear TDT, other B/T-cell markers (including
CD7), and monocyte antigens.
Given these findings, a final diagnosis of ALAL was made, with features of AML and early T-cell precursor
ALL. Given the predominance of myeloid markers, she
was started on AML therapy according to the high-risk
arm of the Children’s Oncology Group AAML1031 study.
She demonstrated significant clinical improvement with this treatment, and bone marrow aspirates and trephines
following the first cycle of definitive therapy revealed
complete morphological and cytogenetic remission, with
a very good partial response on PET-CT and brain MRI.
At the end of treatment, following four cycles of therapy,
she was in morphological, cytogenetic and radiological
remission, but within a month of treatment completion
she presented with pneumonia and left hip joint pain.
MRI revealed multifocal areas of marrow infiltrate in
the lumbar spine, pelvis, and proximal femora, with
diffuse skeletal fluorodeoxyglucose avidity on PET-CT.
An aspirate of a left hip joint effusion was consistent
with disease relapse. An attempt was made to induce
remission using T-cell ALL–based therapy, but she
deteriorated rapidly and died of progressive disease.
DISCUSSION
The World Health Organization has recently updated its classification of ALAL after changes to its definition
over the years.[1] ALAL is uncommon in children and is
particularly rare in infancy. Its exact incidence is difficult
to establish because of changes in definitions over the
years, but accounts for approximately 3% of paediatric
leukaemias.[2] There are no series specifically reporting
the clinical features of ALAL, but it has been stated
that fatigue, infections, and bleeding manifestations
are common presentations.[2] Central nervous system
involvement at the time of diagnosis has been reported
in about 20% of children, significantly more often than
in ALL.[3] [4]
Children with ALL typically present with fever,
bleeding, pallor, fatigue, rash, lymphadenopathy
or organomegaly.[5] Bone involvement occurs at
presentation in about one quarter of patients and
radiological investigations usually reveal a diffuse
pattern of bone change such as osteopenia or radiolucent
metaphyseal bands, rather than one or more focal
masses.[6] AML shares many of the clinical features of
ALL. Other common manifestations of AML include
gingival infiltration and leukaemia cutis.[5] Nonetheless
bone involvement in paediatric AML is uncommon
compared with ALL. Reports of focal bone lesions are
sparse in ALL or AML, and this presentation has not been
reported in children with ALAL. Ghodke et al[7] described
an 8-year-old female who presented with swelling of her
right cheek without other symptoms. Imaging revealed
a solitary soft tissue mass in the right maxillary sinus
and erosion of the alveolar process. Blood, bone marrow
and cerebrospinal fluid examination were all normal.
Immunophenotyping of a biopsy revealed both myeloid
and lymphoid commitment, and a diagnosis of bi/mixed
phenotypic blastic haematolymphoid neoplasm was
made, as bone marrow was not involved.[7]
Our patient presented with pain, irritability and
difficulty weight-bearing. CT showed a focal aggressive
metaphyseal lesion. In contrast to most patients with
ALL and AML, no systemic features were present at presentation and blood tests were essentially normal. Early
diagnosis of ALAL can therefore be very challenging
with this type of presentation. The differential diagnosis
of a focal destructive bone lesion in younger children
includes infection, Ewing sarcoma, metastasis (especially
from neuroblastoma), and Langerhans cell histiocytosis,
in addition to haematological malignancies. Because
early diagnosis and treatment of acute leukaemia reduces
morbidity and mortality,[8] a high degree of suspicion
should be maintained in a child who presents with bone
lesions.
In summary, ALAL is a rare form of acute leukaemia that can present with focal bone lesions in children. Our
patient demonstrated the radiographic challenges in
making an early diagnosis of acute leukaemia and serves
as a reminder that this diagnosis should be considered in
all children with destructive bone lesions.
REFERENCES
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ,
Le Beau MM, et al. The 2016 revision to the World Health
Organization classification of myeloid neoplasms and acute
leukemia. Blood. 2016;127:2391-405. Crossref
2. Duffield AS, Weir EG, Borowitz MJ. Acute leukemias of
ambiguous lineage. In: Jaffe ES, Arber DA, Campo E, Harris NL,
Quintanilla-Martinez L, editors. Hematopathology. Philadelphia
(PA): Elsevier; 2017. p 775-82.
3. Gerr H, Zimmermann M, Schrappe M, Dworzak M, Ludwig WD, Bradtke J, et al. Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol. 2010;149:84-92. Crossref
4. Hrusak O, de Haas V, Stancikova J, Vakrmanova B, Janotova I,
Mejstrikova E, et al. International cooperative study identifies
treatment strategy in childhood ambiguous lineage leukemia. Blood
2018;132:264-76. Crossref
5. Lanzkowsky P, Lipton JM, Fish JD, editors. Lanzkowsky’s Manual of Pediatric Hematology and Oncology. Boston (MA): Elsevier; 2016.
6. Sinigaglia R, Gigante C, Bisinella G, Varotto S, Zanesco L, Turra S.
Musculoskeletal manifestations in pediatric acute leukemia. J
Pediatr Orthop. 2008;28:20-8. Crossref
7. Ghodke K, Tembhare P, Patkar N, Subramanian PG, Arora B, Gujral S. A rare extramedullary and extralymphoid presentation of mixed phenotypic blastic hematolymphoid neoplasm: a study
of two cases. Indian J Med Paediatr Oncol. 2017;38:394-7. Crossref
8. Dang-Tan T, Franco EL. Diagnosis delays in childhood cancer: a review. Cancer. 2007;110:703-13. Crossref