Late-onset Urea Cycle Deficiency is an Under-recognised Cause of Metabolic Childhood Encephalopathy: a Case Report
CASE REPORT
Late-onset Urea Cycle Deficiency is an Under-recognised Cause of Metabolic Childhood Encephalopathy: a Case Report
C Tsoi1, EKC Law1, J Hui2, WCW Chu1
1 Department of Imaging and Interventional Radiology, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong
2 Department of Paediatrics, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong
Correspondence: Prof WCW Chu, Department of Imaging and Interventional Radiology, Prince of Wales Hospital, The Chinese
University of Hong Kong, Hong Kong. Email: winniechu@cuhk.edu.hk
Submitted: 12 Feb 2019; Accepted: 24 Mar 2020.
Contributors: All authors designed the study and acquired the data. CT, EKCL, WCWC analysed the data, drafted the manuscript, and critically
revised the manuscript for important intellectual content. All authors had full access to the data, contributed to the study, approved the final
version for publication, and take responsibility for its accuracy and integrity.
Conflicts of Interest: All authors have disclosed no conflicts of interest.
Funding/Support: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Data Availability: All data generated or analysed during the present study are available from the corresponding author on reasonable request.
Ethics Approval: The patient was treated in accordance with the tenets of the Declaration of Helsinki. The patient provided written informed consent for all treatments and procedures.
INTRODUCTION
Metabolic disorders of the brain can manifest at any age.
During the neonatal period or early infancy they can be
associated with acute and severe illness. Nonetheless due
to the complex genotypic variability and penetrance, a
subset of inborn errors of metabolism can manifest in
late childhood or even adulthood. This often results
in clinically confounding situations and diagnosis is
difficult. We present the clinical and imaging findings in a
6-year-old girl with late-onset urea cycle defect. The
patient initially presented with lethargy and confusion with
signs of encephalopathy. Peripheral blood demonstrated
an increased plasma ammonia level. Multiparametric
magnetic resonance imaging (MRI) including diffusion-weighted
sequence and spectroscopy provided clues to
the diagnosis but the patient deteriorated quickly from
neurotoxicity of the metabolites. A high index of clinical
suspicion, together with biochemical profile and imaging
assessment are required to ensure early initiation of
appropriate treatment.
CASE REPORT
A 6-year-old Chinese girl presented to the accident and emergency department in January 2016 with confusion
and lethargy. She had a 2-day history of upper respiratory
tract infection and fever. Physical examination revealed
a child who was lethargic, disorientated in time and
space, and unable to recognise her parents. Her medical
history was unremarkable. She was born to healthy
non-consanguineous parents, and her antenatal and
postnatal history was unremarkable apart from a similar
but milder episode of vomiting and lethargy 1 year
earlier, which was attributed to influenza A infection,
followed by an uneventful recovery. After admission
to the paediatric intensive care unit she experienced a
few episodes of seizures and status epilepticus. Lumbar
puncture revealed normal biochemistry (cerebrospinal
fluid glucose 6.2 mmol/L, lactate 2.9 mmol/L, protein
0.17 g/L, white blood cell 14 × 106/L) and was negative
for both bacterial and viral infections on culture
and polymerase chain reaction tests, respectively.
Preliminary biochemistry investigations from blood tests
revealed slightly raised lactate (4.4 mmol/L; normal
range 0.7-2.1 mmol/L), a rising trend of ammonia
(143.8-364 μmol/L within 24 hours; normal
<48 μmol/L) and respiratory alkalosis (pH 7.55). Electroencephalography demonstrated diffuse slow
background without normal sleep potential, suggestive
of encephalopathy. Initial plain computed tomography
(CT) scan of the brain on the day of presentation
(Figure 1a) was unremarkable with preserved grey-white
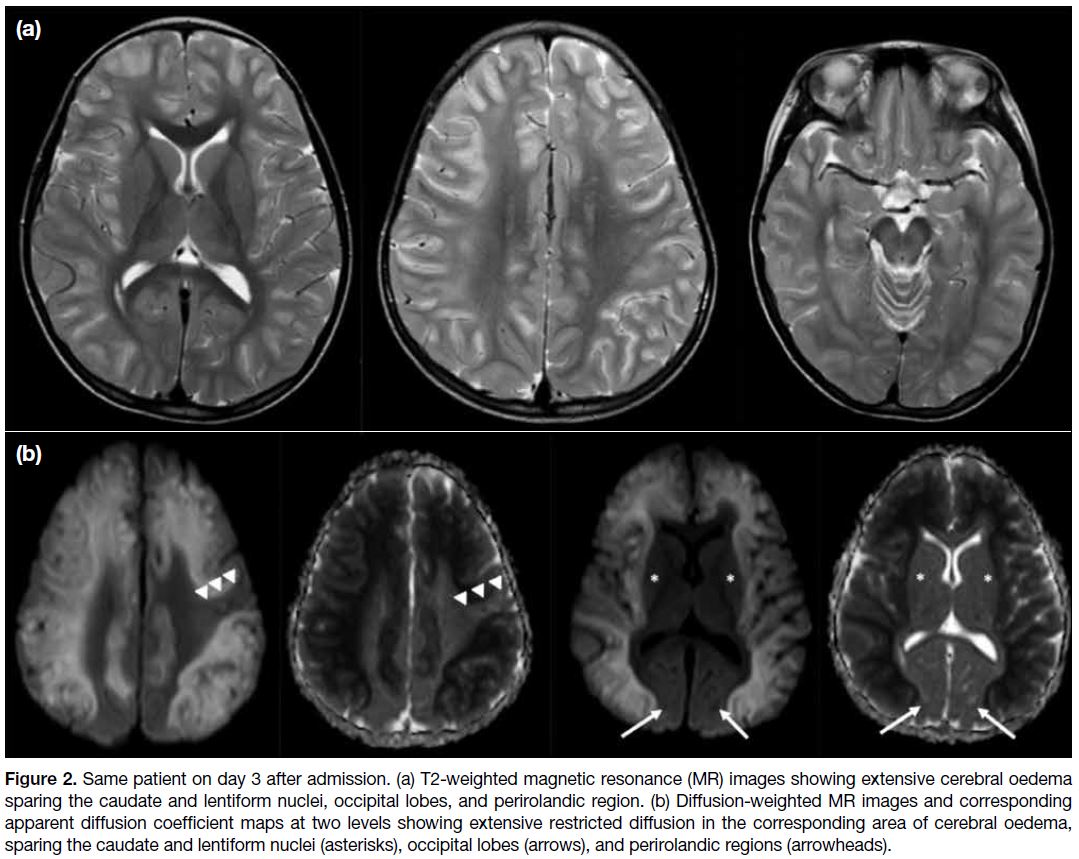
differentiation. MRI brain examination on the
third day of presentation showed extensive T2 signal
changes in both cerebral hemispheres, with restricted
diffusion involving the cortical and subcortical regions
and sparing the perirolandic region, occipital lobes, and
caudate and lentiform nuclei (Figure 2). The dorsomedial
thalami were also symmetrically involved. Follow-up
plain CT scan of the brain (Figure 1b) at 1 day after
MRI examination revealed marked cytotoxic oedema
involving both cerebral hemispheres, in agreement
with earlier MRI findings. Further metabolic screening
showed a markedly raised plasma glutamine level
>1500 μmol/L (normal range, 254-823 μmol/L) and
prompted investigation for urea cycle defect. Further
genetic tests of the CPS1 gene identified a gene mutation
of heterozygous CPS1 c.2407C>G p.(Arg830G1y)
and c.4088_4099de1TGATAGGCATCC. P(Leu1363_I1e366de1), likely pathogenic in nature. Compound
heterozygosity of the two detected likely pathogenic
variants was confirmed by parental genetic analysis.
Results were consistent with a diagnosis of carbamoyl
phosphate synthetase 1 deficiency.
Figure 1. A 6-year-old Chinese
girl presenting with confusion and lethargy. Plain computed tomography
scans of the brain (a) on the day of presentation showing preserved grey-white differentiation, and (b) on day 4 of presentation showing dramatic interval changes with marked cerebral oedema and loss of grey-white differentiation sparing the caudate and lentiform nuclei.
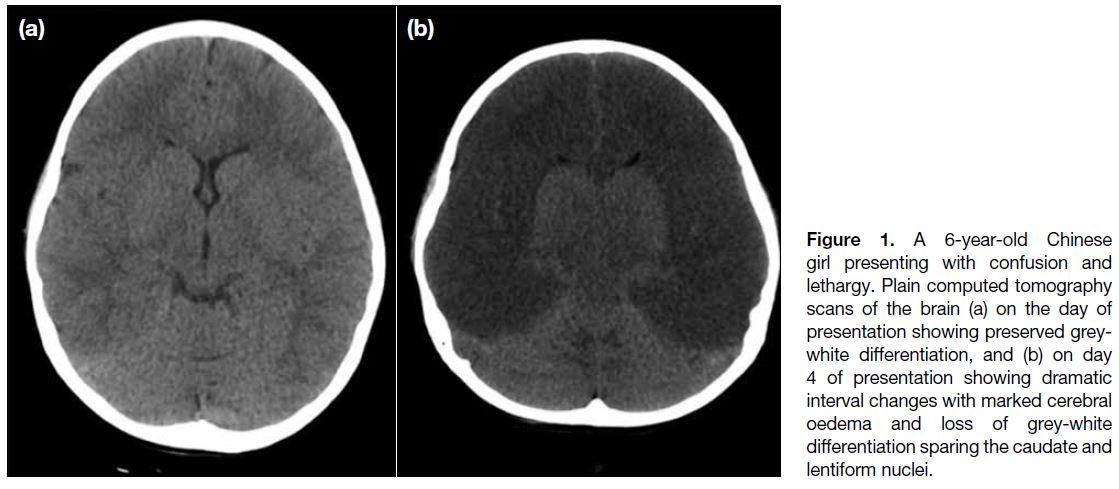
Figure 2. Same patient on day 3 after admission. (a) T2-weighted magnetic resonance (MR) images showing extensive cerebral oedema
sparing the caudate and lentiform nuclei, occipital lobes, and perirolandic region. (b) Diffusion-weighted MR images and corresponding
apparent diffusion coefficient maps at two levels showing extensive restricted diffusion in the corresponding area of cerebral oedema,
sparing the caudate and lentiform nuclei (asterisks), occipital lobes (arrows), and perirolandic regions (arrowheads).
In view of the diffuse cerebral oedema, craniectomy was
performed to relieve the raised intracranial pressure. The
patient was treated with intravenous sodium benzoate/phenylbutyrate infusion to suppress ammonia level and commenced on a low protein, high calorie diet.
Nonetheless subsequent assessment revealed poor
neurological recovery and global developmental delay
due to prolonged hyperammonaemic coma.
Follow-up MRI scan of the brain 6 months later
showed profound diffuse cerebral atrophy and cystic
encephalomalacia with residual white matter change as
sequelae of the previous irreversible insult to bilateral
frontal, anterior parietal and insular regions (Figure 3).
Magnetic resonance (MR) spectroscopy was also
performed on a 3T whole-body scanner (Achieva TX,
Philips Healthcare) using a standard 8-channel head coil.
With image guidance, MR spectroscopy was performed
on a voxel positioned in the right parietal region of the
brain using a PRESS (Point RESolved Spectroscopy)
sequence (repetition/echo times, 2000/31 ms)
with 128 signal averages. Automated second-order
shimming and water suppression were applied prior to
signal acquisition. MR spectroscopy data were exported
and processed offline using LCModel software package
(version 6.3) allowing an accurate measurement of the
metabolites present in the subject. The short echo time
spectrum showed a marked reduction in N-acetylaspartate
(2.02 ppm) related to profound neuronal loss, and
prominent lipid peaks at 0.9 and 1.3 ppm. The presence
of lactate (1.32 ppm) could not be confirmed as the
strong lipid signal at 1.3 ppm found in the spectrum
made the detection of lactate more difficult. Both
choline-containing compounds and glutamine signals
were also elevated, compatible with accumulation of
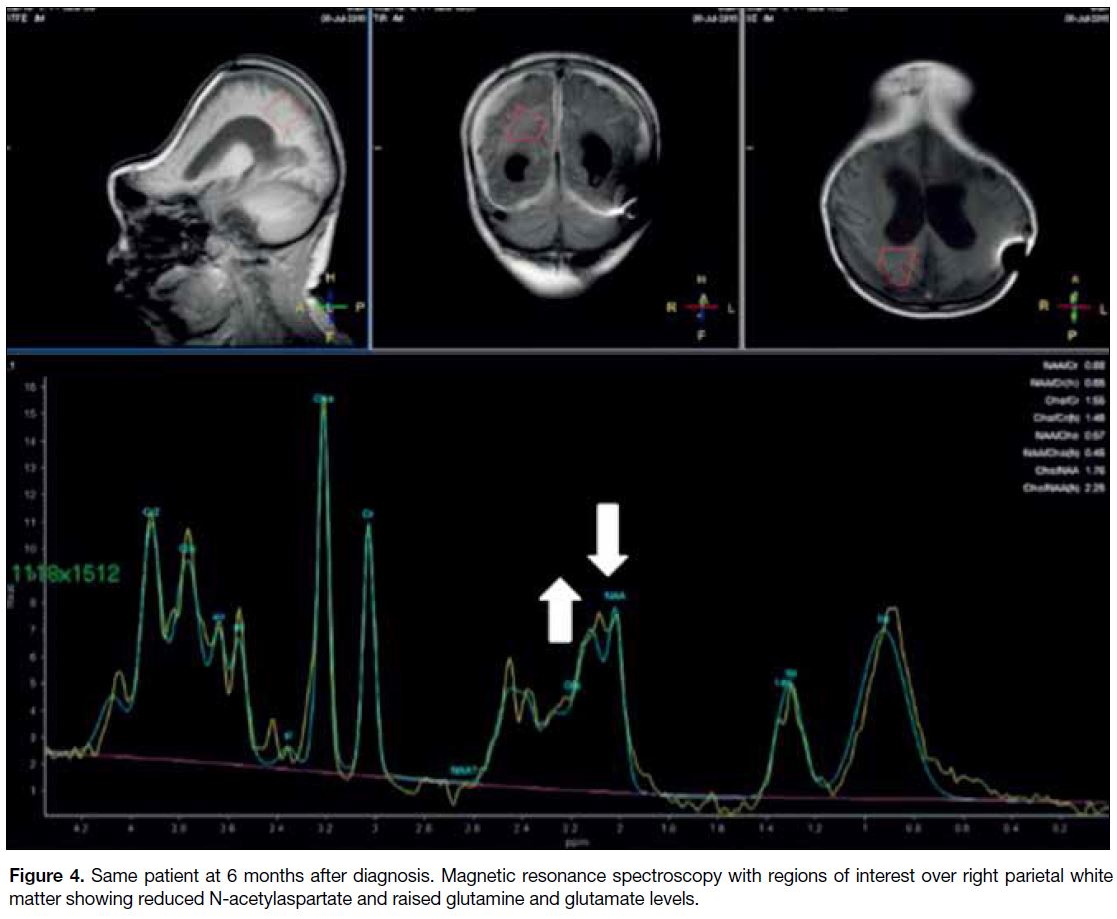
urea precursors (Figure 4).
Figure 3. Same patient at 6 months after diagnosis. (a) Sagittal T1-weighted magnetic resonance (MR) image showing atrophic and
deformed frontal lobe related to previous craniectomy. Fluid attenuated inversion recovery sequences (b) axial and (c) coronal MR images
showing cystic encephalomalacia and residual white matter T2-hyperintensities in bilateral frontal, anterior parietal, and insular regions.
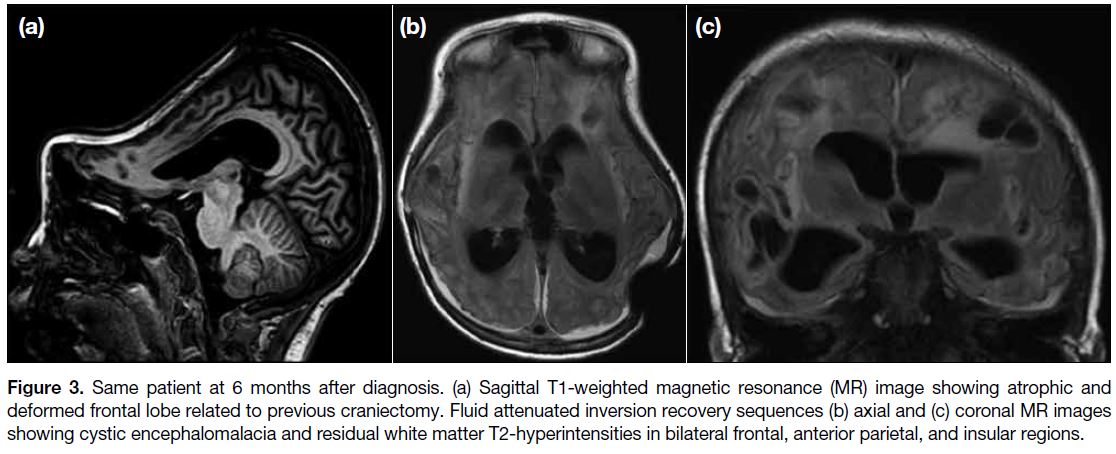
Figure 4. Same patient at 6 months after diagnosis. Magnetic resonance spectroscopy with regions of interest over right parietal white matter showing reduced N-acetylaspartate and raised glutamine and glutamate levels.
DISCUSSION
Childhood encephalopathy is a paediatric emergency
and poses a considerable clinical challenge with a long
list of differential diagnoses. It carries high morbidity
and mortality. Prompt diagnosis and timely treatment
can significantly minimise neurological sequelae in
some cases. The presentation can be acute or insidious.
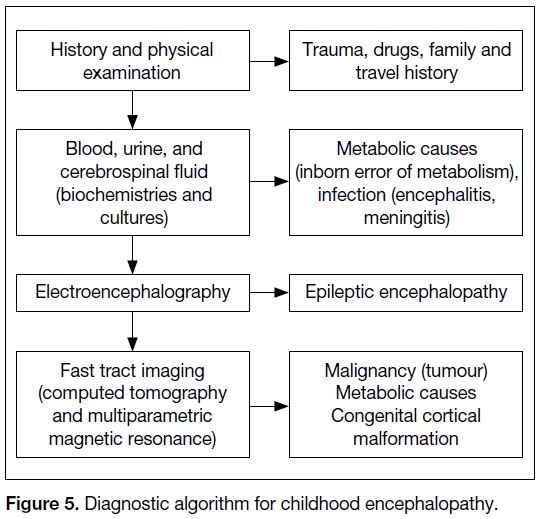
A systematic approach is key to early diagnosis
(Figure 5). In this case, hyperammonaemia was a
hallmark for underlying inborn error of metabolism and
urea cycle defect.
Figure 5. Diagnostic algorithm for childhood encephalopathy.
Urea cycle defect is a group of genetic disorders
associated with deficiency in any one of the five enzymes
involved in the urea cycle.[1] The collective prevalence
of urea cycle defect is approximately 1 in 8000 live
births. Urea cycle defect involves accumulation of urea precursors such as ammonia and nitrogenic compounds
such as glutamine. These chemicals accumulate rapidly
in the brain as ammonia crosses the blood-brain barrier
freely, with a direct consequence of neurotoxicity at a
cellular level, or an indirect effect of glutamine synthesis
that is osmotically active and often leads to cytotoxic
oedema and consequent encephalopathy.[2] Most patients present with neurological symptoms. All defects in the
urea cycle are recessively inherited except for the most
common defect, ornithine transcarbamylase deficiency
that is linked to the X-chromosome.[1]
Patients with urea cycle defects present with a broad
spectrum of symptoms due to variable residual enzyme
activity. Hyperammonaemia crises that include non-specific
symptoms such as loss of appetite, vomiting
and seizures most commonly occur during the neonatal
period. The timing of onset and severity depends on the
nature of the molecular defect and the body’s capacity
to eliminate the toxic nitrogenic products.[2] Affected
neonates often appear healthy for the initial 24 to
48 hours of life, then present with progressive lethargy,
hypothermia, and apnoea. Late-onset patients usually
have a milder form of enzyme defects and often present
from infancy to adulthood with less acute symptoms
such as chronic encephalopathy, learning disorders, and
vomiting. Acute presentation as encephalopathy is often
aggravated by a high protein intake and illness due to
subsequent catabolism that exceeds the body’s threshold
to eliminate toxic precursors.[2]
As illustrated in the current case, our patient was a
late presenter with insidious onset of symptoms. The
metabolic defect was not detected at an early stage of
clinical presentation, leading to a delay in diagnosis
and irreversible and catastrophic neurological damage.
It is worth noting that our patient had presented with a
similar episode of lethargy and vomiting a year prior
to the irreversible insult. The duration and degree of
hyperammonaemia and coma may predict the prognosis
and extent of neurological damage.[2] A high index
of suspicion of a metabolic cause should always be
maintained in cases of neurological derangement since
early treatment may minimise or prevent neurologic
sequelae by minimising protein catabolism and
elimination of ammonia.
Neuroimaging plays an important role in the diagnostic
process.
The initial apparently normal CT brain may have been due to its lower sensitivity to detect acute changes.
From an imaging perspective, the imaging appearances of
different enzyme deficiencies within the urea cycle defect
are similar, likely due to similar metabolic derangements
downstream in the cellular pathway. Hyperammonaemia
and hyperglutaminaemia alter intracellular osmolality,
resulting in brain oedema and intracranial hypertension,
and finally secondary cerebral hypoperfusion.
Similar imaging findings and involvement have been
described previously.[3] [4] Takanashi et al[5] described
five types of MR appearance in urea cycle defects,
speculated to differ by their residual enzyme activity
and clinical severity: (1) extensive diffuse severe
cerebral oedema involving both grey and white matter,
resembling hypoxic-ischaemic encephalopathy (HIE)
in the subacute phase, followed by generalised atrophy,
usually seen in survivors of severe deficiency with
prolonged neonatal coma; (2) infarct-like appearance
in a hemispheric fashion so patients can present with
stroke-like events; (3) less severe ischaemic events
in intervascular boundaries; (4) symmetrical cortical
involvement of the cingulate gyri, temporal lobes,
insular cortex with sparing of perirolandic cortex and
occipital lobes in childhood or adult patients with partial
deficiencies,[3] [4] and (5) preferential involvement of white
matter of the brain, involving the lentiform nuclei, deep
sulci of the insular and perirolandic regions in patients
with neonatal presentation. These areas have highest
metabolic activity at birth and are most susceptible to
hypoperfusion in a hyperammonaemic state, reflected
on MR images and contrast with different patterns of
involvement in children and adults.5 In all cases, it is
speculated that the imaging appearances are related to
underlying hypoperfusion during hyperammonaemia.[5]
The extent of cerebral involvement and presence of
diffusion restriction has a role in predicting the severity
of neurological sequelae.[6]
Among them, type 4 pattern, which was seen in
our case, has also been reported in patients with
hyperammonaemia due to other causes such as valproic
acid-induced hyperammonaemic encephalopathy and
acute hepatic encephalopathy. This suggests that the
appearance might be related to hyperammonaemia
rather than the underlying disorder.[4] This appearance
probably represents a milder form of disease wherein the
ischaemic insults may be partially reversible if promptly
treated. Extensive cortical involvement on MRI (type 1
pattern) mimics severe HIE. Unlike HIE, the degree of apparent diffusion coefficient reduction in urea cycle
defects appears to be less severe and reversible to a
certain degree.[7]
MR spectroscopy provides further information in terms
of the biochemistry. MR spectroscopy appearances
are consistent with the metabolic derangement and
demonstrate elevated glutamine and decreased
myoinositol and choline in severely affected patients.[1]
It can also be used to monitor adequacy of metabolic
control.
CONCLUSION
Recognition of a specific neuroimaging pattern together
with a high index of clinical suspicion of late-onset urea
cycle defect in patients who present with encephalopathy
can guide further genetic tests specific for urea cycle
defect. Early treatment may alleviate and minimise the
devastating neurological sequelae.
REFERENCES
1. Patay Z. MR imaging workup of inborn errors of metabolism
of early postnatal onset. Magn Reson Imaging Clin N Am.
2011;19:733-59. Crossref
2. Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis. 2007;30:865-79. Crossref
3. Bindu PS, Sinha S, Taly AB, Christopher R, Kovoor JM. Cranial MRI in acute hyperammonemic encephalopathy. Pediatr Neurol.
2009;41:139-42. Crossref
4. Takanashi JI, Barkovich AJ, Cheng SF, Kostiner D, Baker JC,
Packman S. Brain MR imaging in acute hyperammonemic
encephalopathy arising from late-onset ornithine transcarbamylase
deficiency. AJNR. Am J Neuroradiol. 2003;24:390-3.
5. Takanashi JI, Barkovich AJ, Cheng SF, Weisiger K, Zlatunich CO,
Mudge C, et al. Brain MR imaging in neonatal hyperammonemic
encephalopathy resulting from proximal urea cycle disorders. AJNR
Am J Neuroradiol. 2003;24:1184-7.
6. Bireley WR, Van Hove JL, Gallagher RC, Fenton LZ. Urea cycle disorders: brain MRI and neurological outcome. Pediatr Radiol.
2012;42:455-62. Crossref
7. Krishna SH, McKinney AM, Lucato LT. Congenital genetic
inborn errors of metabolism presenting as an adult or persisting
into adulthood: neuroimaging in the more common or recognizable
disorders. Semin Ultrasound CT MR. 2014;35:160-91. Crossref